publications
publications by categories in reversed chronological order. generated by jekyll-scholar.
2026
- Preprint
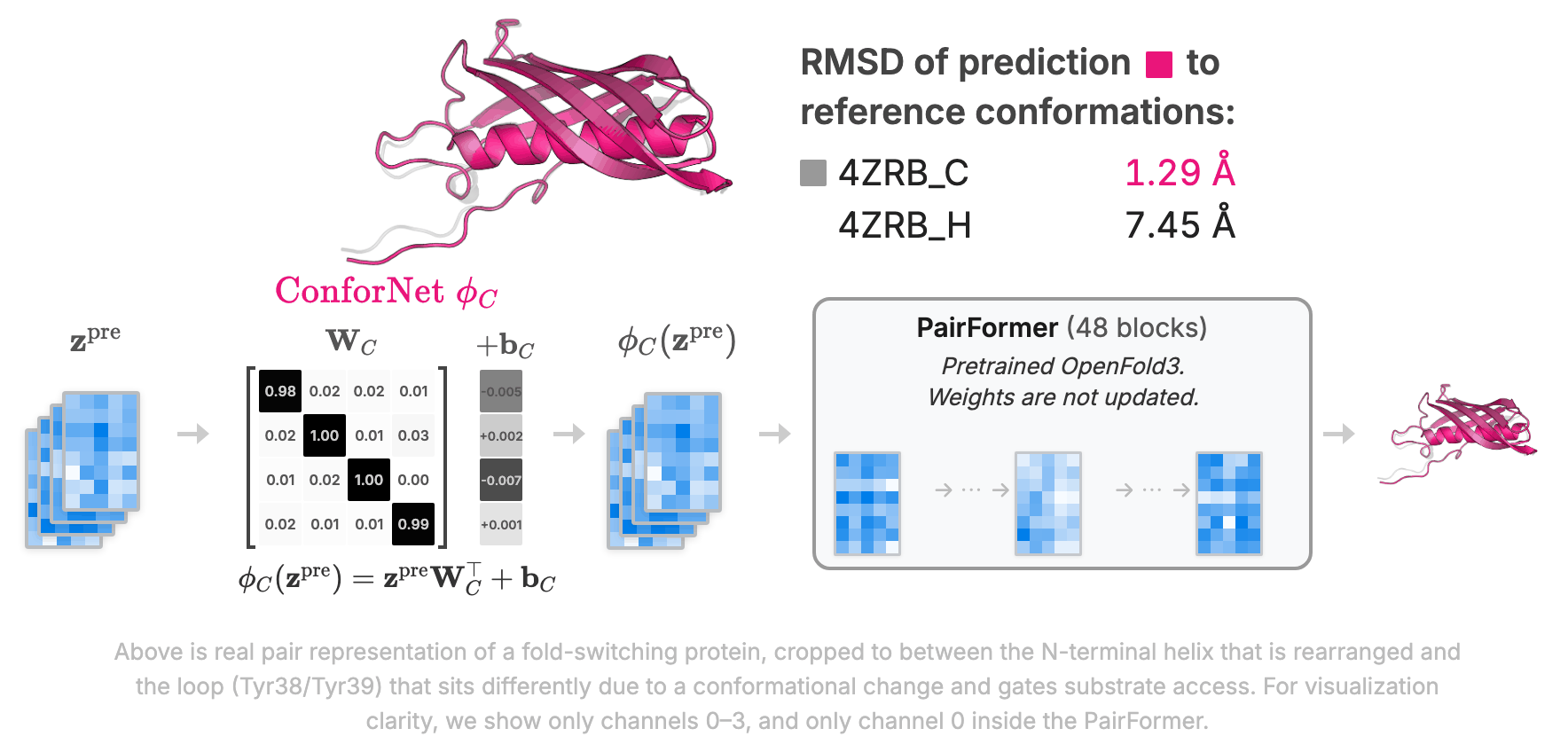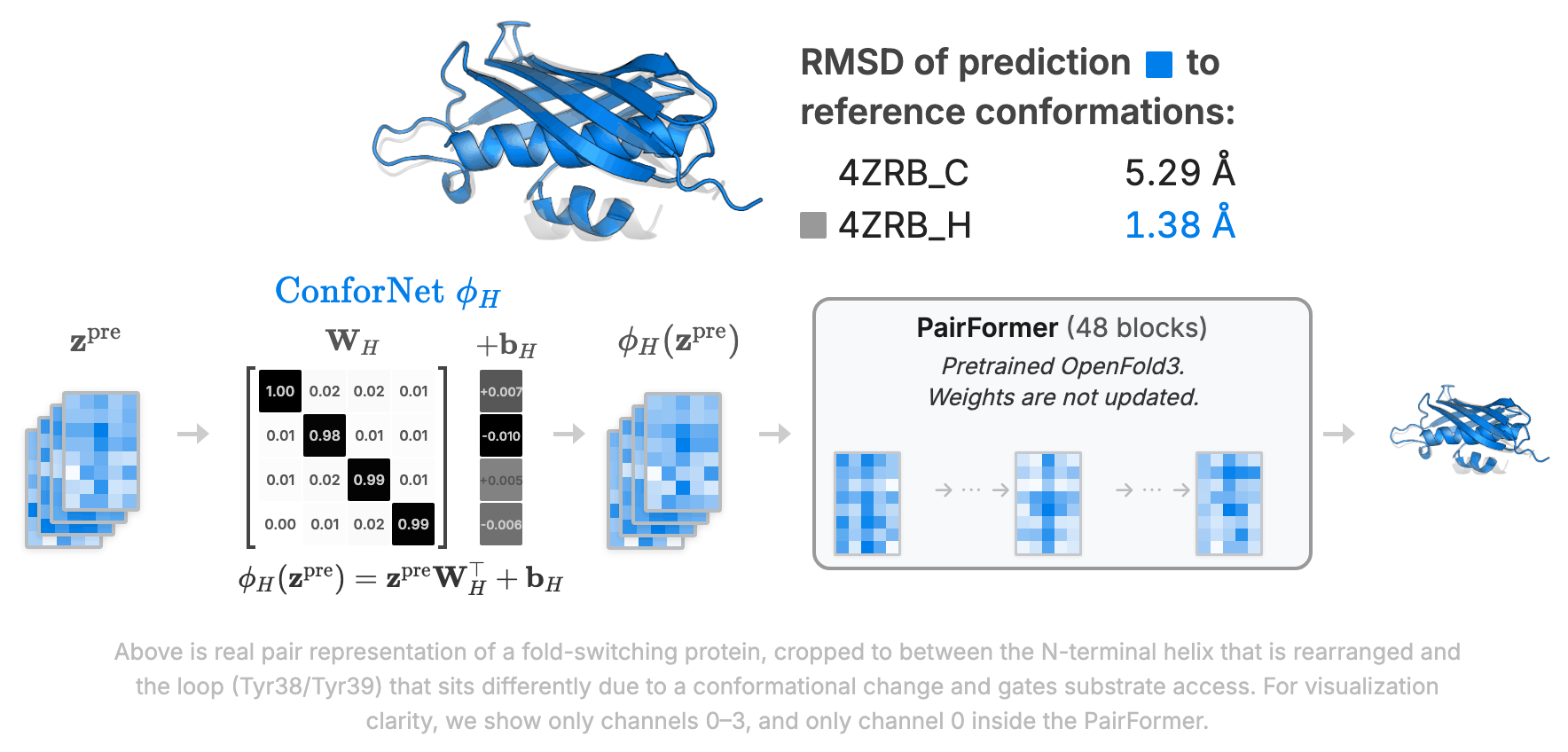 ConforNets: Latents-Based Conformational Control in OpenFold3Minji Lee, Colin Kalicki, Minkyu Jeon, Aymen Qabel, and 2 more authorsarXiv preprint arXiv:2604.18559, 2026
ConforNets: Latents-Based Conformational Control in OpenFold3Minji Lee, Colin Kalicki, Minkyu Jeon, Aymen Qabel, and 2 more authorsarXiv preprint arXiv:2604.18559, 2026Models from the AlphaFold (AF) family reliably predict one dominant conformation for most well-ordered proteins but struggle to capture biologically relevant alternate states. Several efforts have focused on eliciting greater conformational variability through ad hoc inference-time perturbations of AF models or their inputs. Despite their progress, these approaches remain inefficient and fail to consistently recover major conformational modes. Here, we investigate both the optimal location and manner-of-operation for perturbing latent representations in the AF3 architecture. We distill our findings in ConforNets: channel-wise affine transforms of the pre-Pairformer pair latents. Unlike previous methods, ConforNets globally modulate AF3 representations, making them reusable across proteins. On unsupervised generation of alternate states, ConforNets achieve state-of-the-art success rates on all existing multi-state benchmarks. On the novel supervised task of conformational transfer, ConforNets trained on one source protein can induce a conserved conformational change across a protein family. Collectively, these results introduce a mechanism for conformational control in AF3-based models.
- Preprint
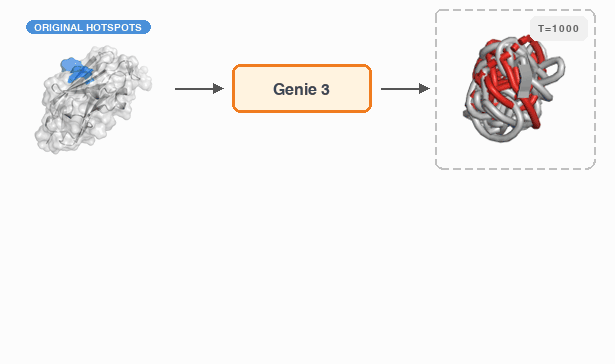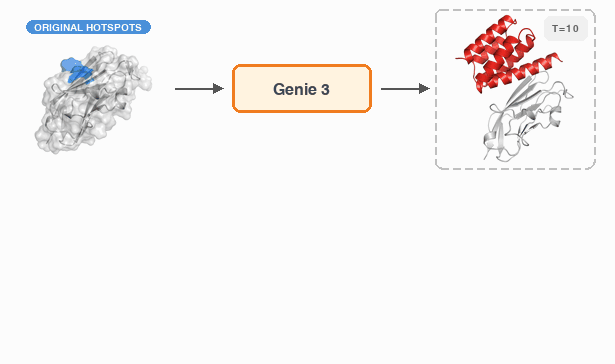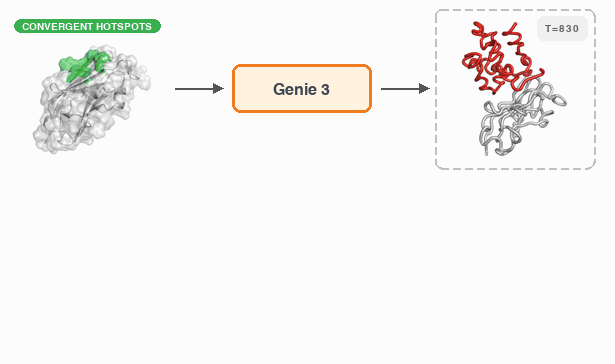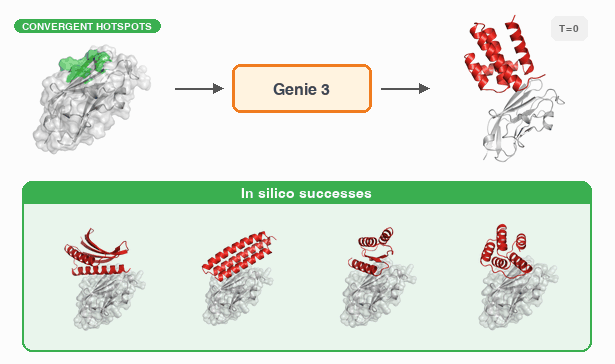 Fast and Ultra-Capable Protein Design: Advancing the Frontier Through Atomistic SE (3)-Equivariance with Genie 3†Yeqing Lin , †Minji Lee, Aakarsh Vermani, Ellena Jiang, and 3 more authorsbioRxiv, 2026
Fast and Ultra-Capable Protein Design: Advancing the Frontier Through Atomistic SE (3)-Equivariance with Genie 3†Yeqing Lin , †Minji Lee, Aakarsh Vermani, Ellena Jiang, and 3 more authorsbioRxiv, 2026Despite the breakneck pace of progress in protein design methodology, frontier problems remain challenging, with leading methods struggling to design high-affinity binders, scaffold multiple functional motifs, or stabilize large multi-domain proteins. Recent research efforts have focused on two areas: improving model reasoning when generating active sites or binding interfaces, and improving concordance between the design process and the in silico oracle used to select promising designs. In addressing the first, the field has shifted towards all-atom models that capture sidechain conformations in atomistic detail by eschewing data-efficient SE(3)-equivariance, mirroring the evolution of AlphaFold2 to AlphaFold3. In addressing the second, recent work has focused on replacing generative models employing diffusion or flow-matching with hallucination approaches that directly optimize the oracle in sequence space; this improves success rates but reduces computational efficiency. Here, we close and surpass the generation-hallucination gap by revisiting SE(3)-equivariance using a branched polymer treatment of protein structures. The resulting diffusion model, Genie 3, achieves state-of-the-art performance on binder design, motif scaffolding, and unconditional generation, while being significantly faster than the best existing methods. We use Genie 3 to design a nanomolar binder of Nipah Glycoprotein G, a tetramer with minimal structural or biophysical characterization, as part of the Adaptyv Bio Nipah Competition, achieving a 12.5% success rate. Taken together, our results present a new frontier in protein design capability and a reexamination of the role of SE(3)-equivariance in molecular modeling.
2025
- ICMLFrom Mechanistic Interpretability to Mechanistic Biology: Training, Evaluating, and Interpreting Sparse Autoencoders on Protein Language ModelsInternational Conference on Machine Learning, 2025
313/12107 = 2.6% of total submissions
Protein language models (pLMs) are powerful predictors of protein structure and function, learning through unsupervised training on millions of protein sequences. pLMs are thought to capture common motifs in protein sequences, but the specifics of pLM features are not well understood. Identifying these features would not only shed light on how pLMs work, but potentially uncover novel protein biology–studying the model to study the biology. Motivated by this, we train sparse autoencoders (SAEs) on the residual stream of a pLM, ESM-2. By characterizing SAE features, we determine that pLMs use a combination of generic features and family-specific features to represent a protein. In addition, we demonstrate how known sequence determinants of properties such as thermostability and subcellular localization can be identified by linear probing of SAE features. For predictive features without known functional associations, we hypothesize their role in unknown mechanisms and provide visualization tools to aid their interpretation. Our study gives a better understanding of the limitations of pLMs, and demonstrates how SAE features can be used to help generate hypotheses for biological mechanisms.
2024
- Preprint
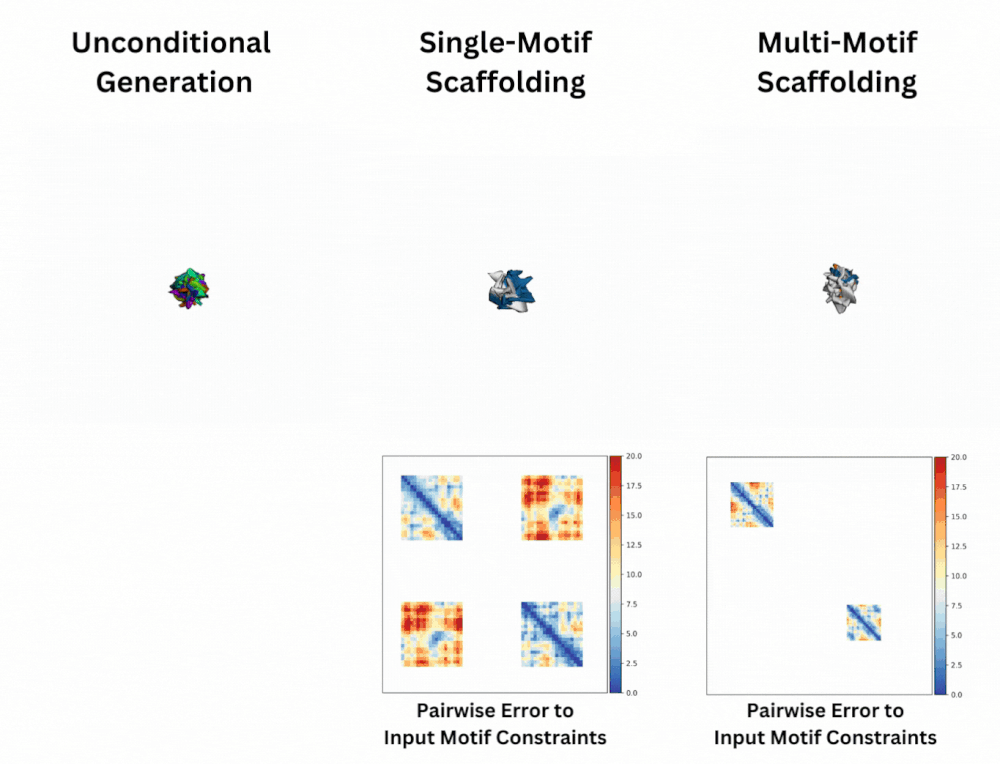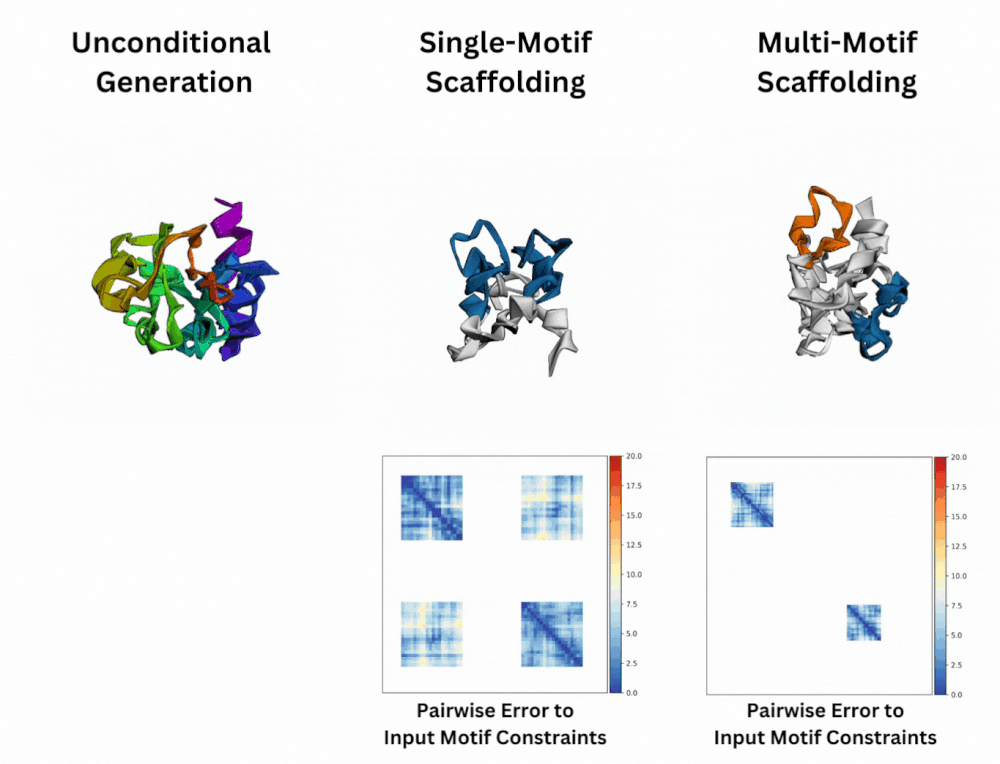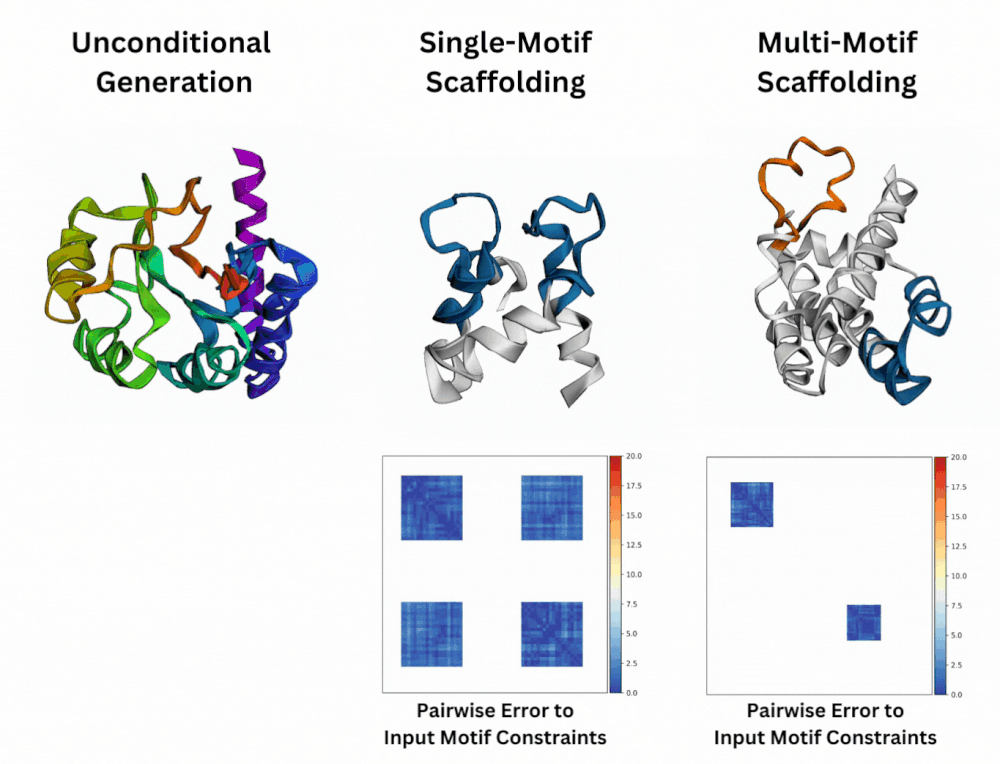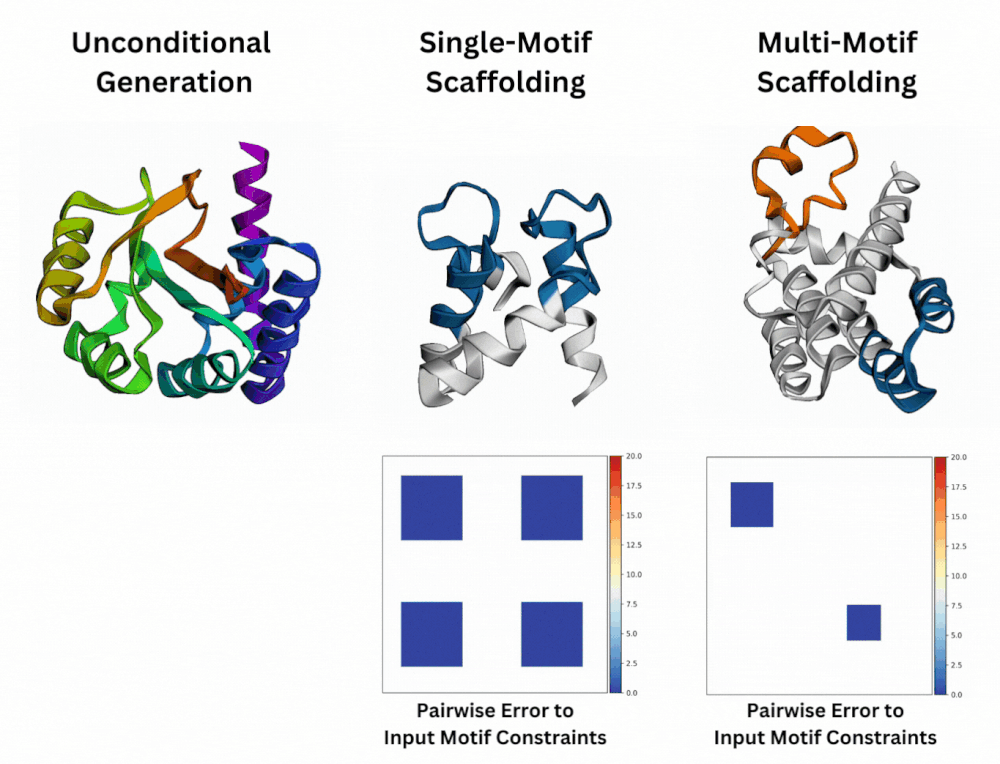 Out of Many, One: Designing and Scaffolding Proteins at the Scale of the Structural Universe with Genie 2Yeqing Lin , Minji Lee, Zhao Zhang, and Mohammed AlQuraishi2024
Out of Many, One: Designing and Scaffolding Proteins at the Scale of the Structural Universe with Genie 2Yeqing Lin , Minji Lee, Zhao Zhang, and Mohammed AlQuraishi2024Protein diffusion models have emerged as a promising approach for protein design. One such pioneering model is Genie, a method that asymmetrically represents protein structures during the forward and backward processes, using simple Gaussian noising for the former and expressive SE(3)-equivariant attention for the latter. In this work we introduce Genie 2, extending Genie to capture a larger and more diverse protein structure space through architectural innovations and massive data augmentation. Genie 2 adds motif scaffolding capabilities via a novel multi-motif framework that designs co-occurring motifs with unspecified inter-motif positions and orientations. This makes possible complex protein designs that engage multiple interaction partners and perform multiple functions. On both unconditional and conditional generation, Genie 2 achieves state-of-the-art performance, outperforming all known methods on key design metrics including designability, diversity, and novelty. Genie 2 also solves more motif scaffolding problems than other methods and does so with more unique and varied solutions. Taken together, these advances set a new standard for structure-based protein design.
- ICML
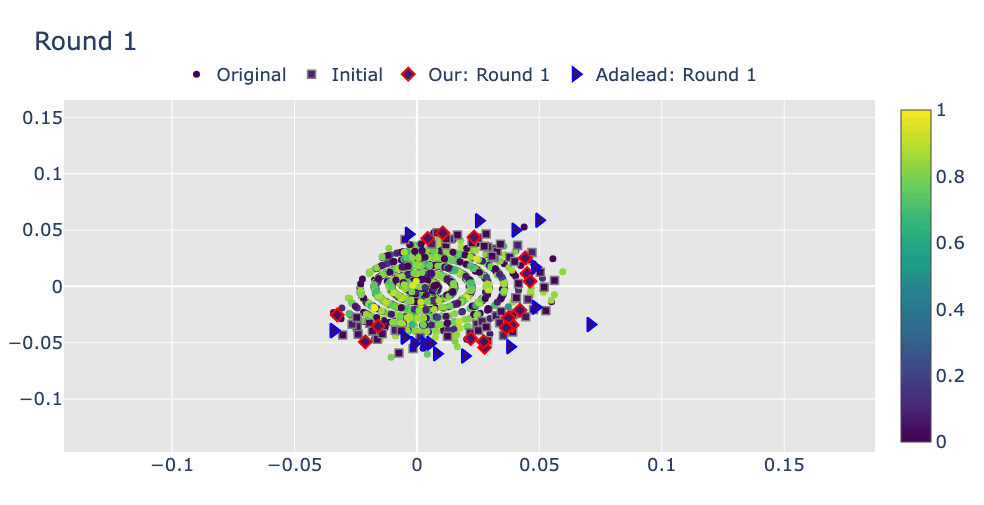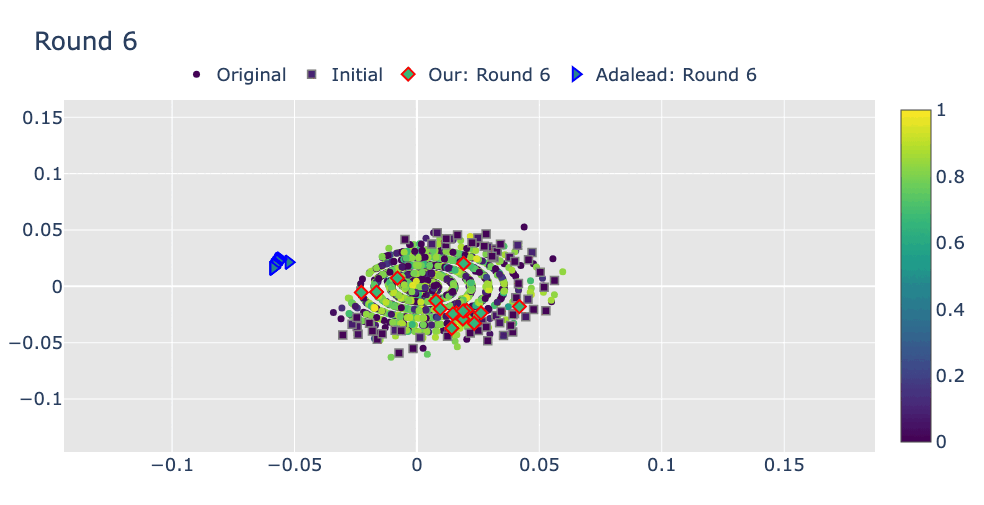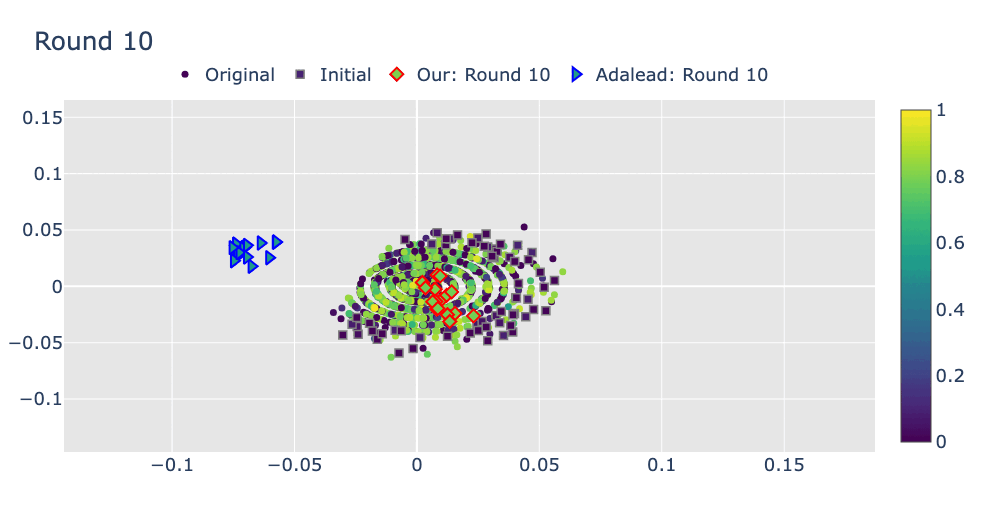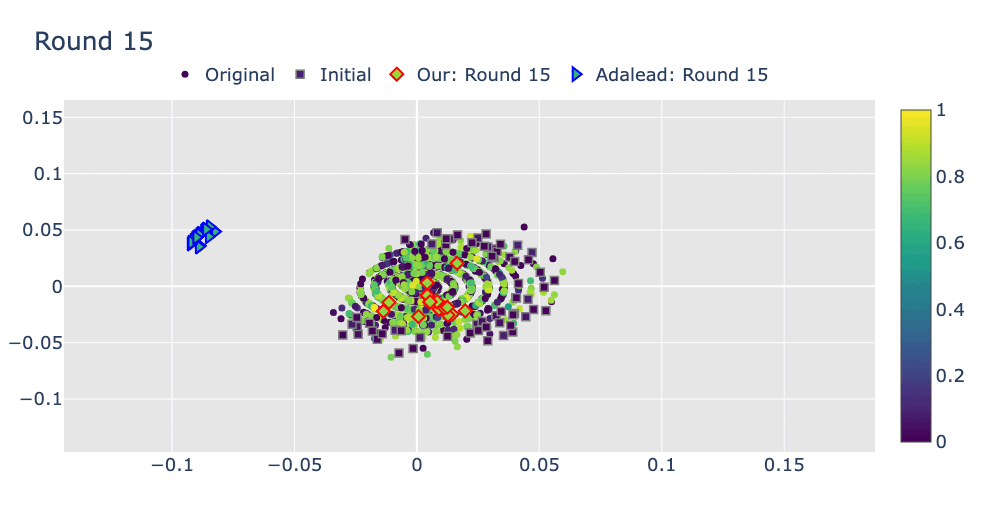 Robust Optimization in Protein Fitness Landscapes Using Reinforcement Learning in Latent Space†Minji Lee, †Luiz Felipe Vecchietti, Hyunkyu Jung, Hyunjoo Ro, and 2 more authorsInternational Conference on Machine Learning, 2024Preliminary version presented at ML in Structural Biology Workshop at NeurIPS, 2022
Robust Optimization in Protein Fitness Landscapes Using Reinforcement Learning in Latent Space†Minji Lee, †Luiz Felipe Vecchietti, Hyunkyu Jung, Hyunjoo Ro, and 2 more authorsInternational Conference on Machine Learning, 2024Preliminary version presented at ML in Structural Biology Workshop at NeurIPS, 2022(144+191)/9473 = 3.5% of total submissions
Proteins are complex molecules responsible for different functions in nature. Enhancing the functionality of proteins and cellular fitness can significantly impact various industries. However, protein optimization using computational methods remains challenging, especially when starting from low-fitness sequences. We propose LatProtRL, an optimization method to efficiently traverse a latent space learned by an encoder-decoder leveraging a large protein language model. To escape local optima, our optimization is modeled as a Markov decision process using reinforcement learning acting directly in latent space. We evaluate our approach on two important fitness optimization tasks, demonstrating its ability to achieve comparable or superior fitness over baseline methods. Our findings and in vitro evaluation show that the generated sequences can reach high-fitness regions, suggesting a substantial potential of LatProtRL in lab-in-the-loop scenarios.
2023
- NeurIPSW
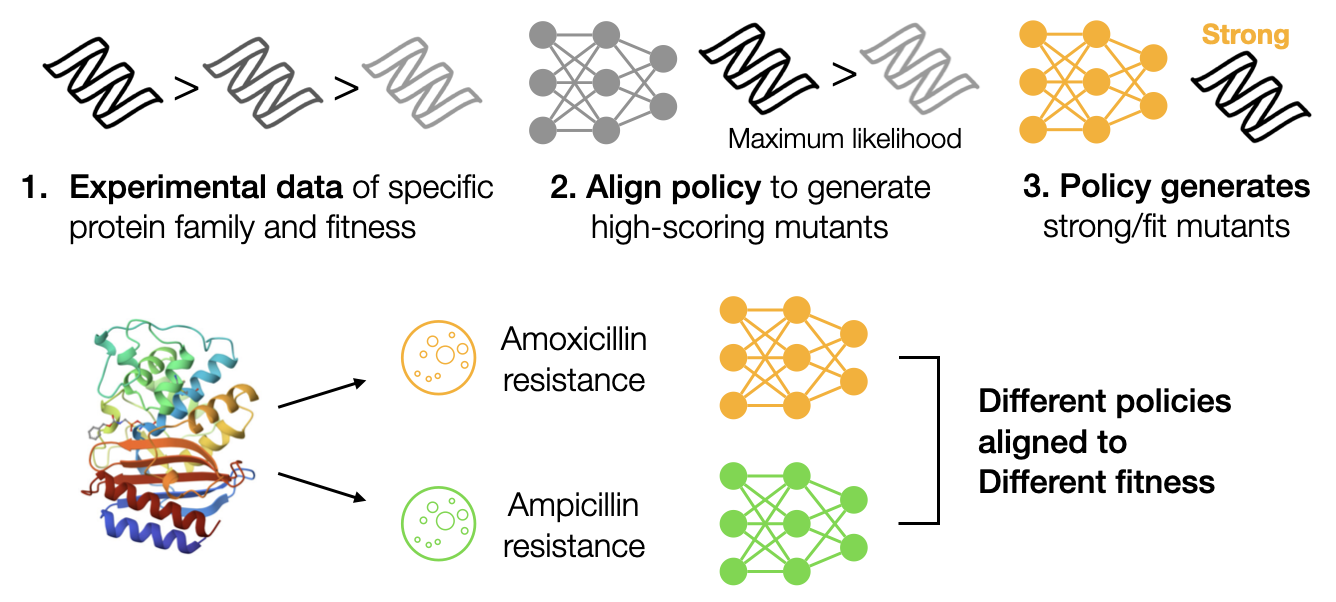 Fine-tuning protein Language Models by ranking protein fitnessMinji Lee, Kyungmin Lee, and Jinwoo ShinGenerative AI and Biology Workshop at NeurIPS, 2023
Fine-tuning protein Language Models by ranking protein fitnessMinji Lee, Kyungmin Lee, and Jinwoo ShinGenerative AI and Biology Workshop at NeurIPS, 2023The self-supervised protein language models (pLMs) have demonstrated significant potential in predicting the impact of mutations on protein function and fitness, which is crucial for protein design. There are approaches to further condition pLM to language or multiple sequence alignment (MSA) to produce a protein of a specific family or function. However, most of those conditioning is too coarse-grained to express the function, and still exhibit a weak correlation to fitness and struggle to generate fit variants. To address this challenge, we propose a fine-tuning framework for pLM to align it to a specific fitness by ranking the mutants. We show that constructing the ranked pairs is crucial in fine-tuning pLMs, where we provide a simple yet effective method to improve fitness prediction across various datasets. Through experiments on ProteinGym, our method shows substantial improvements in the fitness prediction tasks even using less than 200 labeled data. Furthermore, we demonstrate that our approach excels in fitness optimization tasks.
- WACV
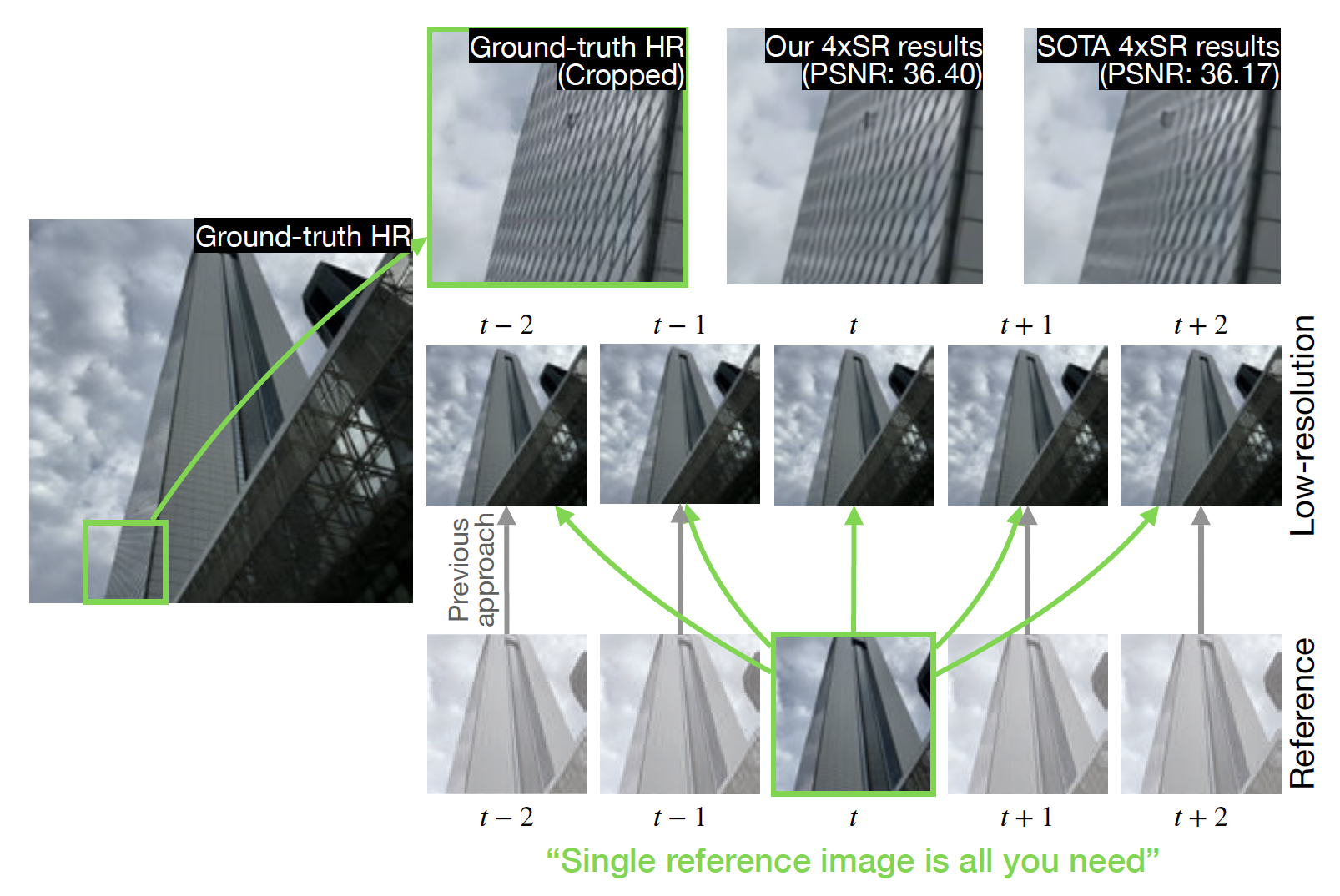 Efficient Reference-Based Video Super-Resolution (ERVSR): Single Reference Image Is All You Need†Minji Lee, †Youngrae Kim, †Jinsu Lim, †Hoonhee Cho, and 3 more authorsIEEE/CVF Winter Conference on Applications of Computer Vision, 2023
Efficient Reference-Based Video Super-Resolution (ERVSR): Single Reference Image Is All You Need†Minji Lee, †Youngrae Kim, †Jinsu Lim, †Hoonhee Cho, and 3 more authorsIEEE/CVF Winter Conference on Applications of Computer Vision, 2023Reference-based video super-resolution (RefVSR) is a promising domain of super-resolution that recovers high-frequency textures of a video using reference video. The multiple cameras with different focal lengths in mobile devices aid recent works in RefVSR, which aim to super-resolve a low-resolution ultra-wide video by utilizing wide-angle videos. Previous works in RefVSR used all reference frames of a Ref video at each time step for the super-resolution of low-resolution videos. However, computation on higher-resolution images increases the runtime and memory consumption, hence hinders the practical application of RefVSR. To solve this problem, we propose an Efficient Reference-based Video Super-Resolution (ERVSR) that exploits a single reference frame to super-resolve whole low-resolution video frames. We introduce an attention-based feature align module and an aggregation upsampling module that attends LR features using the correlation between the reference and LR frames. The proposed ERVSR achieves 12xfaster speed, 1/4 memory consumption than previous state-of-the-art RefVSR networks, and competitive performance on the RealMCVSR dataset while using a single reference image.